-
24 2019-10吉林大学李洋副教授团队:可室温修复化学和机械损伤的超疏水材料
针对以上问题,吉林大学李洋副教授团队以氨基封端聚二甲基硅氧烷(NH2-PDMS-NH2)和异佛尔酮二异氰酸酯(IPDI)为原料合成了可室温自修复的疏水超分子聚合物PDMS-PUa(图1a)。随后将PDMS-PUa,多壁碳纳米管,氯化钠在四氢呋喃中混合,固化成型并除去盐模板,制备出多孔的自修复超疏水材料MCNTs/PDMS-PUa(图1b)。研究结果表明,MCNTs/PDMS-PUa具有良好的超疏水性能,化学稳定性和热稳定性。MCNTs/PDMS-PUa的静态接触角和滚动角分别为153.2°和5.6°,即使在1%的CH3COOH和Na2CO3溶液中浸泡12 h依然保持良好的超疏水性能;此外,在60 ℃条件下放置72 h,MCNTs/PDMS-PUa的多孔结构没有任何塌陷,接触角和滚动角也没有发生变化,保持初始的超疏水性能。
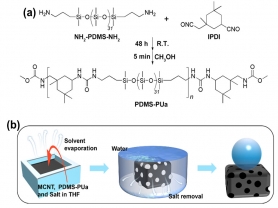
图1. (a) PDMS-PUa的合成路线。(b) MCNTs/PDMS-PUa的制备过程示意图。
由于PDMS-PUa在室温下既具有良好的分子运动能力,因此,当其受到化学腐蚀而导致其超疏水性能丧失后,MCNTs/PDMS-PUa内部的PDMS-PUa分子在自由能的驱动下扩散到受损的表面,恢复材料原始的表面物质组成及超疏水性能(图2a)。如图2b所示,当受到等离子刻蚀的MCNTs/PDMS-PUa在室温条件下储存放置48小时之后,MCNTs/PDMS-PUa表面的水的静态接触角从0°恢复到150.9°,滚动角也恢复到6.8°,证明MCNTs/PDMS-PUa在室温下修复化学损伤的能力。更重要的是,由于PDMS-PUa聚合物分子间氢键的动态可逆性,当两个损伤的断面相互接触时,断面PDMS-PUa聚合物分子之间再次形成氢键从而修复受损的MCNTs/PDMS-PUa (图2c)。如图2d所示,当把切为两半MCNTs/PDMS-PUa重新接触在一起并在室温下储存放置72 h,MCNTs/PDMS-PUa重新连接在一起并且可以承受自身的重量。切痕修复区域的接触角和滚动角分别恢复到153.1°和5.9°。
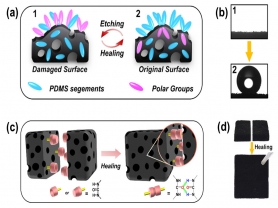
图2. (a) MCNTs/PDMS-PUa修复化学损伤的示意图。(b) MCNTs/PDMS-PUa室温修复化学损伤前后的接触角。 (c) MCNTs/PDMS-PUa修复机械损伤的示意图。(d) MCNTs/PDMS-PUa室温修复机械损伤前后的照片。
由于其稳定的多孔结构和超疏水性能,MCNTs/PDMS-PUa可以用于清除水表面漂浮的油污。此外,得益于MCNTs/PDMS-PUa的自修复能力,MCNTs/PDMS-PUa可以在受到化学腐蚀和穿刺后依然保持高效的油水分离能力。
以上相关成果以“Superhydrophobic Foams with Chemical- and Mechanical-Damage-Healing Abilities Enabled by Self-Healing Polymers”为题发表在《ACS Appl. Mater. Interfaces》上。论文的通讯作者为李洋副教授。
论文链接:
https://pubs.acs.org/doi/10.1021/acsami.9b11858
-
23 2019-09哈佛大学锁志刚教授课题组:瞬时强韧非共价粘接
自然界中普遍存在着两种材料通过非共价键粘合的过程——非共价粘接,其在很多基本生命过程中起到了重要作用,如基因复制、蛋白质折叠、细胞移动等。相比共价粘接,非共价粘接可在室温下快速形成,且不受催化剂、温度等条件的限制。因此,便捷的非共价粘接在日常生活中有着广泛应用,例如压敏胶、药物传递、伤口缝合等。然而,现有方法并未充分发挥非共价粘接的潜能:实现与共价粘接相当的强韧粘接。导致这一结果的根本原因在于缺乏理论基础,顶层设计方法的缺失极大阻碍了非共价粘接方法的进一步发展和应用。
近日,哈佛大学锁志刚教授课题组提出了通过选择不同类型非共价键作为增韧因子和互联因子实现瞬时强韧粘接的基本原理,并在短短数秒内使粘接能达到750 J/m2以上。在该研究中,研究人员以拥有共价键交联聚合物网络和非共价键增韧因子的水凝胶通过非共价键互联因子与另一材料粘接为例,将力学与化学相结合,从理论和实验两方面揭示了增韧因子与互联因子的强弱关系、材料性能以及环境因素(pH、温度等)对非共价粘接行为的影响,证明了所提原理的正确性与普适性,为非共价粘接的进一步发展与应用奠定了理论基础。另外,研究人员还开发出了具有不同拓扑结构的非共价粘接应用形式,包括胶带、粉末、分子魔术贴、胶水和分子粘合环。同时展示了其在水下粘接、可逆粘接、导电粘接、可分离粘接等工程和生物医学领域的应用前景。
基本原理
如图1所示,当一个外力试图分离粘接在基底上的水凝胶时(含有共价键网络和非共价键增韧因子),水凝胶的共价键网络将力传递到界面分离处的裂纹尖端,产生应力集中,从而造成互联因子的开裂。这个过程可以被形象地理解为解开拉链的过程。如果互联因子的形成过程是瞬间的,那么宏观粘接过程也将瞬间形成。如果互联因子足够强韧以致很多增韧因子开裂,那么粘接也将会是强韧的。
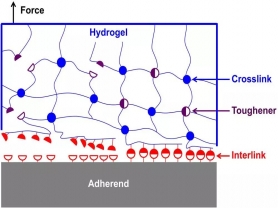
图1 瞬时强韧的非共价粘接基本原理
瞬间形成的非共价粘接
首先,研究人员对非共价键互联因子进行系统研究后发现:尽管单个非共价键互联因子相对于共价键互联因子较弱,但当非共价键互联因子的数量足够多时,其强韧度可比肩共价键互联因子。研究人员通过氢键(PAA的羧基与PAAm的氨基间形成)相互作用将聚丙烯酸(PAA)水凝胶与聚丙烯酰胺(PAAm)水凝胶粘接在一起。粘接的强弱可通过90度剥离实验测得的粘接能表征。如图2A所示,两界面接触30秒内粘接能便超过了160 J/m2,接近于拥有共价交联网络水凝胶的韧性。这里,PAA的pH为1.5,PAAm的pH为3.5。当PAA和PAAm贴合在一起后,氢离子和水分子会在二者之间扩散,直到达到平衡。因此,粘接能随着接触时间的增加而先降低、然后稳定。当PAA和PAAm的pH同为3.5时,粘接能便不再随接触时间而变化(图2B)。
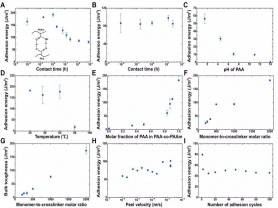
图2 系列因素对PAA-PAAm粘接能的影响
由于PAA和PAAm之间是通过羧基与氨基之间形成的氢键粘合在一起的,其粘接能依赖于pH。考虑到丙烯酸的室温解离常数为4.5,pH>4.5时,多数羧基电离,形成较少的氢键,粘接能降低;pH<4.5时,更多的羧基会参与氢键形成,粘接能升高(图2C)。另外,粘接能还与温度有关,当温度升高时,氢键会越来越容易被破坏。实验表明,当温度升高到80 ℃时,粘接能会急剧下降(图2D)。当PAA同PAA-co-PAAm共聚水凝胶通过氢键粘合在一起时,其粘接能会随着PAA在共聚物中所占比例的增加而提高(图2E),从而说明氢键数目越多,粘接能越高。
研究人员还发现,PAA与PAAm的粘接能以及PAA自身的韧性都会随着PAA水凝胶交联度的降低而升高(图2F和G)。同时,其粘接能还和剥离的速度有关。当剥离速度接近零时,PAA与PAAm的粘接能仍然接近60 J/m2(图2H)。进一步验证了PAA和PAAm之间是通过稳定且强韧的氢键、而非物理缠绕的方式粘合在一起的。
相比共价粘接,非共价粘接的独特优势在于其粘接过程的可逆性。研究人员将PAA和PAAm周期性地贴合与剥离,发现其粘接能在第一个贴合-剥离周期后有所下降,然后在接下来的100个周期内保持稳定且较高的水平(图2I)。
除了PAAm之外,PAA还可以通过氢键与其他水凝胶、生物组织以及弹性体瞬间形成强力的粘接(图3)。其中,研究人员对弹性体的表面行了等离子处理,让弹性体表面富含羟基,从而与PAA中的羧基形成氢键,产生粘接。
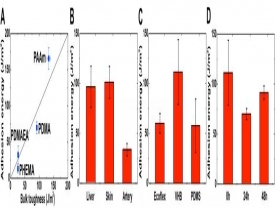
图3 PAA与各种材料间的快速非共价粘接
瞬时强韧的非共价粘接
接下来,为了表明非共价粘接可以既快速又强韧,研究人员分别向PAA和PAAm中引入非交联的PAA长链和钙-海藻酸钠(Ca-alginate)复合物作为增韧因子,同时增强了PAA和PAAm(图4A)。为了进一步增强互联因子,alginate-PAAm韧性水凝胶的pH被设定为4.5。如图4B 所示,粘接可以在30秒内快速形成,且粘接能超过了750 J/m2。
为了研究增韧因子与互联因子的强弱关系对粘接强弱的影响,研究人员通过化学设计对二者的强度逐一进行调节,并测量了相应的粘接能。首先,保持互联因子强度不变(即保持非交联PAA长链的量不变),通过钙浓度的变化改变增韧因子的强度。实验发现,当钙的浓度较低时,粘接能与没有加入增韧因子时的PAA与PAAm的粘接能相当。而当钙的浓度较高时,增韧因子过强,因而不会开裂,所以粘接能也不高(图4C)。其次,保持增韧因子强度不变(即保持钙的量不变),研究人员通过改变非交联PAA长链的量来改变互联因子的强度并研究了粘接能的变化。结果表明,PAA长链越多,互联因子越强,因而粘接能越高(图4D)。
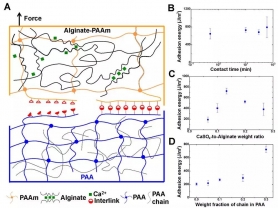
图4 瞬间形成的强韧非共价粘接
通过上述研究,如图1 所示的瞬时强韧非共价粘接的基本原理得到了验证,并且具有普适性。当互联因子瞬间形成时,非共价粘接的形成将会是瞬间的。当互联因子足够强以致于很多增韧因子开裂时,非共价粘接可以和共价粘接一样强韧。
多种拓扑结构的非共价粘接形式
在现实生活中,不同的应用场景需要不同的粘接形式。以PAA为例,研究人员开发了具有不同拓扑结构的非共价粘接应用形式,包括胶带、粉末、分子魔术贴、胶水和分子粘合环(图5)。每一种形式的PAA都可以与另外一种材料通过氢键瞬间形成强韧的非共价粘接。
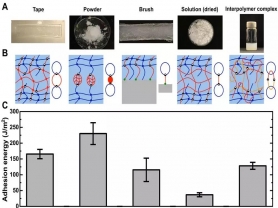
图5 多种拓扑结构的非共价粘接
在不破坏材料的情况下将两个强力粘接在一起的物体分离极具挑战性。这里, 研究人员通过分别向两种强力粘合材料间界面滴碱溶液和热水的方式,实现了pH分离和热分离,且未对材料本身造成任何破坏。
另外,PAA作为透明且可拉伸的离子导体,可用于导电粘接。被切成两段的含盐PAAm可拉伸离子导体通过PAA被重新连接在一起,从而恢复其导电性能和可拉伸性能。除此之外,研究人员还实现了水下粘接。
总结与展望
该研究提出了瞬间形成强韧非共价粘接的基本原理:合理选择不同非共价键互联因子与非共价键增韧因子。瞬间形成的强韧非共价粘接可以拥有多种拓扑结构的粘接应用形式,包括胶带、粉末、分子魔术贴、胶水和分子粘合环。此外,非共价键类别的多样性为实现水下粘接、导电粘接、可逆粘接以及可分离粘接等多种功能提供了极大的设计空间。
该研究工作发表于ACS Applied Materials & Interfaces。论文第一作者为王叶成博士(哈佛大学博士、哈佛大学博士后),第二作者为贾坤教授(西安交通大学)。其他合作者包括项春平(西安交通大学博士在读、哈佛大学访问学生)、杨加伟博士(哈佛大学博士、麻省理工学院博士后)及姚晰教授(河南大学)。哈佛大学、美国科学院与工程院院士锁志刚教授为论文通讯作者。
论文链接:
https://pubs.acs.org/doi/10.1021/acsami.9b10995?from=groupmessage&isappinstalled=0
-
31 2019-08ICD 失效的常见机理简析
ICD失效,即 Inner connection defects,又叫内层互连缺陷。对于PCB生产厂家而言,ICD问题在电测工序较难有效拦截,往往是流到下游甚至是客户端,在进行SMT贴装过程,PCB板经历无铅回流焊IR、波峰焊接、以及一些手工焊或是返修等高温制程的冲击下,发生内层互联失效开路,而此时的PCB板已进行了组装,因而会产生极大的品质风险。
下面简要从钻孔质量和除胶过程这两个方面,阐述 ICD 失效的影响机理,对此类问题的检测和分析经验进行小结。
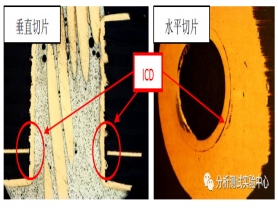

>>>>钻孔质量对 ICD 的影响
以高频高速材料的加工为例,高频高速板材的基板具有低Dk 、低Df 的特性,其极性小,材料活性低,去除胶渣困难,从而进一步加大了孔内残胶对PTH电镀的影响。部分高频板材介质层树脂填料多,物理特性比较硬,对钻刀的钻嘴磨耗大,孔壁粗糙度大,以及钻嘴摩擦高温凝胶较多。因此,在孔壁粗糙度大、钉头异常等情况可能导致除胶药水循环不良,造成除胶效果不佳,未能完全将孔壁残留胶渣去除干净,导致内层孔铜与孔壁连接处产生ICD失效。也有一部分板材的基材偏软且软化点低,钻孔加工过程中产生的高温易使钻屑软化、粘附成团,造成入钻排屑不畅而形成间歇性的挤出排屑,钻屑易被挤压粘附在孔壁上,极大的增加了后工序除胶处理难度,存在孔内残胶风险,最终可能会导致ICD问题。

对于刚-挠结合印制板(FPC),柔性材料的绝缘介质为聚酰亚胺(PI)等,这些材料的机械加工性能相对较差,加工过程中产生形变大,容易导致钉头产生,也就在内层互连的位置留下了应力,因此,这种情况下钻污残留对孔铜和内层铜层的互连可靠性的影响会更加突出,最终导致产品在受到焊接过程的高温冲击时,内层互连处裂开而出现开路。

1 案例背景
>>>>除胶不净对 ICD 的影响
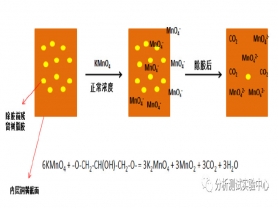
以普通环氧树脂体系的FR-4板材为例,在化学除胶过程中,钻孔后残留在孔壁的高分子环氧树脂胶所包含的C-O、C-H等化学键可被KMnO4氧化分解,生成对应的二氧化碳和水等无机物,再经水洗或除油后,可被完全清除干净。当出现KMnO4浓度低于控制限范围等异常情况时,孔内壁的基铜层就可能会残留树脂胶,经除胶、水洗、除油等流程后均无法有效除净,电镀时,在有胶的区域基铜层与电镀铜结合力较差,经热应力处理后,残胶区域受到热应力的“拉扯”而出现微裂纹现象。

对于高频材料而言,其介质层与钻刀的钻嘴磨损更剧烈,钻嘴摩擦高温导致凝胶过多,因此,高频板的除胶过程需更加注意,有些产品甚至采用“等离子除胶+化学除胶”两次除胶相结合的流程,来确保孔壁残胶被完全清除,防止残留的胶渣造成内层铜环与电镀孔铜之间结合不良,从而导致ICD失效。
参考资料文献:
[1] 张军杰, 朱雪晴等.高频基材混压印制板的ICD问题探讨[J] .印制电路信息.2018,NO.12:59—63.
[2] 钟岳松,刘再平.浅谈刚挠印制板内层互连失效与钻孔参数的关联[J] . 印制电路信息. 2018,NO.02:32—35.
-
31 2019-08失效分析案例:激光盲孔底部裂纹失效案例分析
1 背景描述
电子设计在不断提高整机性能的同时,也在努力缩小其尺寸。从手机到智能武器的小型便携式产品中,"轻、薄、小"是永远不变的追求。而PCB制造工艺中的高密度集成(HDI)技术可以使终端产品设计更加小型化,同时满足电子性能和效率的更高标准。HDI技术目前广泛应用于手机、数码(摄)像机、笔记本电脑、汽车电子和其他数码产品等,其中以手机的应用最为广泛。

采用激光盲孔作为主要的微导通孔是HDI关键技术之一。激光盲孔孔径小而孔数多的特点是实现HDI板高布线密度的有效途径,因此,激光盲孔的可靠性直接决定到产品的可靠性。从业内现有的研究成果来看,激光盲孔的可靠性主要取决于制程工艺流程和介质层材料。一般来说,引起盲孔失效的主要原因包括:(1)由于盲孔孔铜与底铜的结合力不良,产品在使用过程中盲孔孔铜与底铜出现分离;(2)由于盲孔脚部孔铜较薄,产品在使用过程中盲孔脚部孔铜断裂。本文以一例HDI产品激光盲孔底部裂纹失效案例为切入点,讨论了盲孔与底铜结合力不良的失效机理。
2 盲孔底部裂纹案例
分析
2.1 化铜层针孔导致盲孔底部裂纹
2.1.1 案例背景
某型号的光电通讯设备在可靠性测试过程中的高温和低温状态时,均发现整机不能稳定工作。经最终排查确认,是PCBA的激光盲孔电阻值出现异常,失效现象为:常温下,故障网络上测得的阻值为367.68 mΩ;将PCBA放入125℃恒温条件下的高温试验箱内部一段时间后,阻值逐渐增大至882.65mΩ,最终发生开路故障。再从125℃的恒温条件冷却至室温后,故障网络上的阻值为395.41mΩ。
2.1.2 故障定位
对故障网络施加一个大小为0.4A的直流恒定电流,根据焦耳定律Q = I2*R*t可知,阻值偏大的区域发热量也越大。使用红外热成像仪对故障网络进行发热量探测,根据PCBA上发热量高低的分布情况,来定位出阻值偏大的点,分析结果如下图2所示:
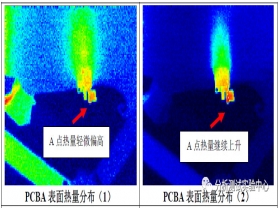
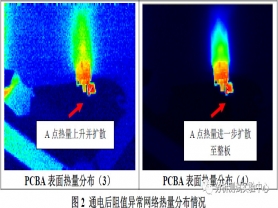
从上图2中可知,随着恒电流通电时间越长,故障网络上焊接点A处发热量最大,表观温度最高,而且随通电时间延长,热量逐渐上升,并扩散至全板,说明A点处阻值偏大,且有可能是导致故障网络电阻值不稳定的主要原因。
2.1.3切片分析
通过微切片法对A点位置进行分析,经离子研磨抛光处理后,使用扫描电镜SEM对其金相观察,分析结果如下图3所示:

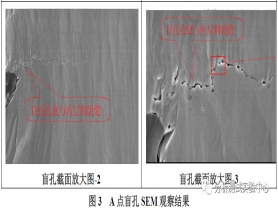
通过上述切片图3可以明显的观察到,A点盲孔底部存在微裂缝,裂缝发生在化学沉铜层界面,微裂缝的存在导致盲孔与底部的机械埋孔之间接触不良。当PCBA在高温下受热时,基材膨胀,使得裂纹逐渐扩大,从而导致电阻值逐渐增大,直至开路的现象。激光盲孔的制作流程为:激光钻孔→等离子→化学清洗→AOI扫描→沉铜→电镀填孔,盲孔底部的裂缝发生在沉铜层与内部机械埋孔的结合界面。
2.1.4 机械拔孔试验及盲孔底部观察
将盲孔中的孔铜直接拉拔出来,使用扫描电镜观察盲孔底部和孔铜底部的情况,分析结果如下图4所示:



由图4中对盲孔孔铜底部的SEM观察可知,拔孔过程所造成的断裂界面为盲孔孔铜与内层铜的结合界面,这说明盲孔的孔铜与内层铜之间结合力较弱。另外,盲孔底部的化学沉铜层表面存在有大量针孔,导致盲孔孔铜与内层铜面之间的有效结合面积减小,影响结合力。因此,在后期的热处理过程中,PCB基材受热膨胀产生内应力,而盲孔孔铜与内层铜面的结合力较弱,盲孔被拉扯导致出现裂缝的失效现象。
2.1.5小结
激光盲孔与内层铜之间产生裂纹的原因主要是由于孔底化铜层存在针孔缺陷,导致盲孔孔铜和内层铜结合强度减弱,不能抵抗多次回流高温所带来的冲击,从而造成盲孔孔铜与内层铜之间被拉裂。
2.2 孔底余胶导致盲孔底部裂纹
盲孔在制备过程中需经过激光钻孔、等离子等关键处理过程,其中激光钻孔的主要作用是将盲孔内的树脂烧蚀,但在此过程中可能由于激光钻孔能量不足或树脂的返沉积作用,盲孔底部有少量的树脂残留,在后续的等离子除胶或化学除胶处理过程中,处理不充分,会导致盲孔底部基铜上有余胶,造成盲孔与内层铜层之间结合力不足。
2.2.1切片分析
对激光盲孔位置进行切片分析,分析结果如图5所示,可见盲孔底部存在裂缝。

2.2.2盲孔底部分析
将盲孔拉拔出来,对其底部进一步分析,发现存在黑色的物质,类似树脂胶渣,如图6。对黑色物质进行元素分析,其含有C、O、Si、Ca和Br元素,为树脂成分。因此,盲孔底部存在树脂胶渣,导致盲孔与基铜的结合力较弱,在后期的热处理过程中,基材受热发生膨胀,盲孔被拉扯从而出现底部裂纹的现象。
2.2.3小结
在电镀填孔流程前,盲孔底部存在有少量余胶,导致盲孔孔铜与底部铜面之间的结合力较弱,在后期的热处理过程中,基材受热发生膨胀,盲孔孔铜与底部铜面的结合力较弱,盲孔被拉扯导致出现裂缝
3 结论
(1) HDI板激光盲孔可靠性失效的现象常表现为线路网络中阻值不稳定或导通不良,且阻值受温变的影响较大,因此,在常规的电测试工序,很难进行有效检测和拦截;
(2) 激光盲孔底部化铜针孔缺陷或孔底余胶等品质不良均会造成盲孔底部产生裂纹开路,导致产品出现可靠性失效。
-
31 2019-08北京大学李星国 J Mater Chem A:等离子体与MOF的“碰撞”制备高分散性析氢催化剂

【引言】金属有机骨架材料(Metal organic frameworks,MOFs) 是是由金属离子和有机配体组装而成的新型三维多孔材料,其规整性及多样性等特性为设计各种需求的催化剂提供了一个理想的平台。通过高温热处理是去除MOFs中配体部分将其转化为相关衍生材料(氧化物,碳包覆金属等)的是目前制备纳米催化材料的一种有效策略,至今,以 MOFs 为前驱体/牺牲模板,已经成功制备了大量具有复杂组成、结构和功能的纳米结构催化剂。
然而,大多数工作的热转化条件往往需要非常高的温度(600 ~900 ℃),这样的高温热处理过程不仅比较耗能,而且热解过程中金属原子极易发生团聚,导致催化剂颗粒变大,其中的活性位点的密度因金属的团聚而减小,阻碍了反应物与部分活性位点的接触,无法充分发挥材料的催化活性。
【成果简介】
基于此,近日,北京大学李星国教授和郑捷副教授课题组发展了一种新型的氨气气氛下低温等离子体对MOFs材料的改性策略,将具有高化学活性的 NH3 plasma引入对Ni基MOF的处理过程中,得到氮掺杂寡层碳包覆的超微镍纳米颗粒(Ni@NC)作为高性能氢析出反应(HER)催化剂,并对NH3 plasma与MOFs材料的相互作用机制进行了研究。
NH3等离子体的引入既可通过化学刻蚀作用降低大大降低 MOFs的分解温度,同时等离子体中的含氮活性物种(Reactive nitrogen species, RNS)可促进配体的快速碳化,极大地抑制了纳米颗粒的迁移和团聚现象,得到具有“杨桃型”特征形貌的超微 Ni 纳米颗粒(2~3 nm),使金属位点充分暴露,提高纳米结构的催化活性,在 10 mA cm-2 电流密度下HER过电势仅为 61 mV。同时,该 Ni@NC 催化剂可应用于微型的太阳能-氢能能量转换系统中,成功在大电流密度下持续产生氢气,有望实现以太阳能为能量来源耦合电解水技术持续产氢的实际应用。相关成果以题为“Plasma modification of a Ni based metal–organic framework forefficient hydrogen evolution”的文章发表于J. Mater. Chem. A上,第一作者为北京大学郭妍如博士。
【图文导读】
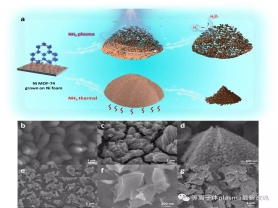
图1 a) NH3气氛等离子体与NH3气氛直接热解处理Ni MOF-74示意图;不同样品的SEM图:b)Ni MOF-74,c~d) NH3-plasma样品,e~f)NH3-thermal样品,g)Ar-plasma样品

图2 样品的TEM和HRTEM图:a, d, e) NH3-plasma样品,b)NH3-thermal样品,c, f)Ar-plasma样品
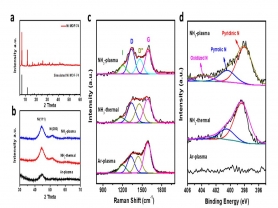
图3 a)前驱体Ni MOF-74的XRD图;b~d)不同条件处理样品的表征:b)XRD图,c)Raman图谱;d)N 1s图谱
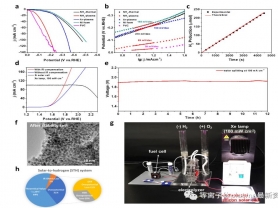
图4 样品电化学性能测试:a)LSV曲线;b)Tafel斜率;c)法拉第效率;d)与商用Si太阳能电池联用的两电极全电解性能(NH3-plasma样品为HER催化剂,光强:AM1.5,100 mW cm-2);e)全电解测试稳定性;f)稳定性测试后样品TEM图;g)微型的太阳能-氢能能量转换系统实物照片;h)系统能量损失图
【小结】
等离子体技术作为一项集物理学、化学、生物学和环境科学于一体的交叉综合性技术,近些年来在材料制备和改性方面产生了很多新兴应用研究,如对材料进行表面处理、掺杂、缺陷制造等,该研究工作将等离子体与对金属有机骨架的改性联系起来,碰撞出新的火花,为MOFs材料的修饰和应用开辟了新的思路。同时,高度化学反应性和低热效应的组合使得低温等离子体有望成为开发高性能催化剂的非常有前途的技术。
Guo, Y.; Gao, X.; Zhang, C.; Wu, Y.;Chang, X.; Wang, T.; Zheng, X.; Du, A.; Wang, B.; Zheng, J.; Ostrikov, K.; Li,X. Plasma modification of a Ni based metal–organic framework for efficienthydrogen evolution. Journal of Materials Chemistry A. 2019, 7, 8129-8135.
DOI:10.1039/C9TA00696F
-
31 2019-08天大刘昌俊教授ACS Catal:Plasma在催化剂合成中的应用
前言:
在催化剂的发展过程中,各种新催化剂合成技术和工艺相继出现,比如原子层沉积,光还原法等。自上世纪九十年代起,等离子体作为一种催化剂的制备工艺被越来越受到关注。近期天津大学的刘昌俊教授课题组综述了近些年来Plasma在催化剂合成上的应用(ACS Catal. 2018, 8 (3), 2093-2110)。本文旨在与大家分享其中的主要内容。

什么是等离子体?
所谓“等离子体(Plasma)”即处于电离状态的气体。等离子体由大量的电子、离子、中性原子、激发态原子、光子和自由基等组成。当然等离子体也符合电中性原则,也就是电子和正离子的电荷数相等,这就是“等离子体”的含义。一般来说,等离子体是利用高电压将一种气体或气体混合物电离形成。根据体系能量状态、温度和离子密度,等离子体通常可以分为高温(high-temperature)等离子体和低温(low-temperature)等离子体。低温等离子体又包括热等离子体(thermal)和冷(nonthermal/cold)等离子体。在Plasma的影响下,催化剂合成过程中的成核、生长等过程会明显区别于传统的热合成过程。在Plasma中,某些热力学不利的过程可能会很容易发生,一些低温低压下难以合成的相态在室温下就有可能被合成。
利用不同的将能量耦合到Plasma的手段,使用不同电极或者介电材料,可以得到各种不同类型的Plasma。比如,direct current (dc)、 alternative current (ac) glow discharges、 radio frequency (rf) discharges、microwave discharges、dielectric barrier discharges (DBD)、gliding arcs、arcs、plasma jets 和plasma torches。目前Plasma在催化剂工艺中已经被成功用于催化剂的还原(reduction)、氧化(oxidation)、掺杂(doping)、刻蚀(etching)、合金化(alloying)、表面处理(surface treatment)和净化(surface cleaning)方面(如图1)。文献中详细总结了Plasma是如何在催化剂合成中起作用的。
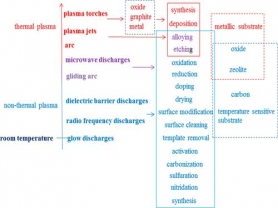
图1. 不同的Plasma类型及在催化剂合成中的常见应用。
1. 还原(Plasma reduction)
还原催化剂中的金属离子是Plasma最常见的用途。Plasma中的电子,氢自由基等还原性组分能够直接将催化剂前驱体中的金属离子还原从而得到相应的金属催化剂。比如利用非氢辉光放电作为电子源就能产生室温电子,这些电子可以有效用于金属离子的还原。这种低温的电子还原相对于高温还原法,金属成核更快而生长却相对更慢,这样在控制催化剂颗粒尺寸方面具有明显的优势。该方法非常适合将贵金属颗粒负载在有序介孔材料中,并且可以获得尺寸可控的贵金属纳米线(图2)。

图2.利用室温Plasma合成的Au纳米线。
室温电子还原也被证明能够有效用于核壳结构双金属催化剂的合成(如Au@Pd, Pd@Pt)。除了增加催化剂活性中心的分散性之外,冷Plasma能够促进催化剂活性组分的表面富集。比如Di课题组利用室温Plasma处理Pd/FeOx可以将Pd富集在催化剂表面。[2]
2. 氧化和降解(Plasma oxidation and decomposition)
2.1 除去催化剂上的有机配体、模板和污染物(Plasma Removal of Organic Ligands, Template, and Contaminants)
PVP等有机大分子在纳米催化剂或多孔材料的合成中经常被用作稳定剂、溶剂、表面活性剂或模板等。无论是作为多孔材料模板剂还是纳米颗粒的保护剂,这些大分子在催化应用中都需要除去。但这些有机配体或大分子的去除并不容易,比如常用的热处理法在模板剂的除去过程中常会导致多孔材料的塌陷。相对来说,冷离子体提供了一种低温移除有机配体的途径,冷离子体不仅能降解材料表面的有机分子也能够渗透入孔道内部起作用。目前为止,冷Plasma已经被成功用于有机配体CTAB,催化剂表面石墨碳层,制备多孔材料的碳模板,P123模板,合成沸石的结构导向剂氢氧化四丙基铵等的有效去除。
2.2 氧化(Plasma oxidation)
Plasma氧化也可以在低温下进行,这样就可以避免热氧化过程中可能导致的各种问题(如烧结等)。Plasma氧化可以用于催化剂积碳后的再生。当然最直接的,其可用于金属催化剂的表面氧化。低温Plasma曾被用于Cu foil, Ag foil和Au film的表面氧化,氧化后的催化剂被证明能有效促进CO2电还原为CO。
2.3 分解(Plasma decomposition)
催化剂前驱体(硝酸盐等)的分解是很多催化剂制备中的基本步骤。一般来说,前驱体分解是通过氧气或空气氛围中的热处理/煅烧来实现的。这个过程中,前驱体的分解,挥发和相变同时发生。Plasma处理可以作为替代热处理的良好选择。热Plasma和非热Plasma都可以用于催化剂前驱体的分解。热Plasma可以认为是非热Plasma和热处理过程的结合。当然,热Plasma处理过程的时间不能太长,否则同样会导致烧结等问题,因此更多的文献报道集中在冷Plasma降解催化剂前驱体。
3. 喷涂(Plasma spray)
Plasma喷涂是一种利用热Plasma的制备工艺。该工艺以dc arc和rf plasma来产生高温的离子化气体作为喷射源,催化剂前驱体通过惰性气体带入离子流从而被加温和反应。之后plasma流被喷向基底材料,催化剂前驱体以颗粒形式沉积在基底上。Plasma喷涂可以方便地合成具有多层“催化层/保护层”的复合催化材料。
利用Plasma喷涂法,Abatzoglous课题组报道合成了用于F-T合成反应的Co/C和Fe/C催化剂。[3-5]这些催化剂表现出远优于浸渍法和沉积沉淀法合成的Co/C和Fe/C的催化活性。Plasma喷涂法也同样可以制备合金类催化剂。比如,Gulyaev课题组合成了PdCeC复合物,经过高温除碳得到的PdCeOx能有效催化CO的氧化反应。在室温条件下,其活性高于化学法制备催化剂的2-3倍。[6]
4. 沉积(Plasma deposition)
Plasma沉积包括直接Plasma沉积,Plasma促进的原子层沉积(PEALD), Plasma促进的化学气相沉积(PECVD)等。根据催化剂前驱体和沉积过程的不同,热Plasma或冷Plasma都可能被用于沉积过程。
热Plasma最重要的应用之一就是将金属、金属氧化物等化合物沉积在多孔材料中。操作中,沉积金属作为阴极并在阴极上蒸发,加速然后沉积到基底上。该方法的优点在于可利用金属或石墨做阴极将催化剂直接沉积在载体材料上,从而避免了使用不同催化剂前驱体或者是结构导向剂对催化剂造成的影响。更重要的是,Plasma沉积法的重复性非常高,非常适合用于研究催化剂的本征活性。文献中报道中已经涉及了Plasma沉积法制备各种金属、金属氧化物催化剂,并且此类催化剂已经成功用于CO氧化,氧还原、CO2甲烷化和电解水等催化反应中。
5. Plasma合成硫化物、氮化物、磷化物等特定化合物(Plasma synthesis of sulfides, nitrides, phosphides, and other specific compounds)
近些年来,硫化物、氮化物、磷化物等化合物在催化,尤其在电催化方面受到相当的关注。Plasma合成法也被证明是一种合成此类化合物的有效方法。比如,Ouyang课题组利用rf N2 plasma活化技术将泡沫Ni转化成了多级结构三维氮化镍 (hNi3N)并成功用于析氧反应(OER)中。[7]他们用类似方法处理NiMo合金膜制备出的三维多孔NiMoN可以作为析氢反应(HER)的催化剂(图3)。利用含H2S组分的Plasma也可以成功制备诸如CdS,WS2等化合物。[8]
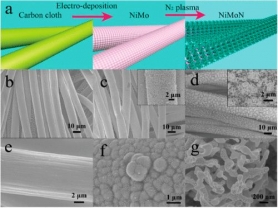
图3.三维多孔NiMoN的制备方法。载体碳布(b,e),电沉积的NiMo合金(c,f)和15min N2plasma处理后的NiMoN催化剂(d,g)的SEM图片。
过渡金属磷化物作为电解水的催化剂被广泛报道。一般情况下磷化物的合成需要较高的温度(> 1000oC),而利用等离子体可以大大降低磷化物的合成温度和磷前驱体的使用量。金属,金属氢氧化物,金属氧化物都可以直接在低温(100-300 oC)下迅速(30s-20min)被磷化为相应的磷化物。除了常见的氮化物、硫化物和磷化物以外,plasma还能用于合成一些其他低温下难以合成的特定化合物,比如炭基材料,氟化物和硼化物等。
6. 表面处理(Surface treatment)
冷Plasma已经被广泛用于对催化剂表面进行处理和改性。这些处理包括制造表面空穴、表面缺陷、表面掺杂、表面粗糙化、改性表面基团等。比如,Bharti课题组利用简单的air plasma处理Fe-和Co-doped TiO2薄膜能够在TiO2表面产生丰富的Ti3+和氧空穴。[9] Tripathi和Islam利用氧plasma处理P-type硅基底上的Fe催化剂,能够明显增加碳纳米管生长的成核位点,并且能够改变碳纳米管的生长方向。[10] Yang课题组发现,氧plasma处理后的Al2O3担载的Fe催化剂比未处理的Al2O3担载的Fe具有更好的抗烧结性能, 因此能够得到尺寸更长的碳纳米管。[11] Plasma也是一种高效地向碳材料中引入缺陷和表面官能团的方法。它能够除去炭基材料中不稳定的sp3碳和无定型的sp2碳,从而能暴露出更多的有效活性位点。比如利用air rf plasma对热解法得到的Fe-N/C催化剂进行处理,可以暴露更多的FeN4活性位点,从而提高催化剂的氧还原活性。当然,不仅对表面的处理,Plasma可以对催化剂的整体结构进行刻蚀和掺杂,综述的第10部分也列举了很多例子,而作用原理是类似的。
7. 辅助合成含有软/温度敏感型载体的催化剂(Plasma preparation with soft and temperature-sensitive supporting materials)
因孔道发达,基团丰富等特点,多孔有机材料、导电聚合物、高比表面碳材料、多孔凝胶和多孔分子筛材料常被用于金属催化剂的载体材料。但是这些材料耐高热性能差,以致难以使用热还原的方法还原其中的金属组分。低温Plasma提供了一种完美的解决方式。除此之外,冷plasma能够促进多种有机单体的高速聚合形成固体聚合物。结合plasma促进聚合和还原的两种特性,可以很容易合成“高分散的金属/聚合物”类型的复合材料。
综上所述,等离子体在催化剂的合成方面提供了一种有效途径,相对于传统的溶液化学法和热处理等方法,冷离子体表现出一些独特的优势。其能有效地解决催化剂合成的大部分挑战。相信作为一种相对较年轻的方法,等离子体在催化方面会受到更多关注并且一些新的相关工艺和方法也会出现。有方法有兴趣的同学可以关注这篇综述论文。
参考文献:
[1]. Wang, Z.; Zhang, Y.; Neyts, E. C.; Cao, X.; Zhang,X.; Jang, B. W. L.; Liu, C.-j., ACS Catal.2018, 8 (3), 2093-2110.
[2]. Di, L. B.; Li, Z.; Park, D.; Lee, B.; Zhang, X. Jpn. J.Appl. Phys. 2017, 56, 060301.
[3]. Blanchard, J.; Abatzoglou, N.; Eslahpazir-Esfandabadi,R.; Gitzhofer, F. Ind. Eng. Chem. Res. 2010, 49, 6948-6955.
[4]. Aluha, J.; Boahene, P.; Dalai, A.; Hu, Y.; Bere, K.;Braidy, N.; Abatzoglou, N. Ind. Eng. Chem. Res. 2015, 54, 10661-10674.
[5]. Aluha, J.; Abatzoglou, N. Gold Bull. 2017, 50, 147-162.
[6]. Gulyaev, R. V.; Slavinskaya, E. M.; Novopashin, S. A.;Smovzh, D. V.; Zaikovskii, A. V.; Osadchii, D. Y.; Bulavchenko, O. A.; Korenev,S. V.; Boronin, A. I. Appl. Catal., B 2014, 147, 132-143.
[7]. Ouyang, B.; Zhang, Y.; Zhang, Z.; Fan, H. J.; Rawat, R.S. Small 2017, 13, 1604265.
[8]. Zhang, Y.; Ouyang, B.; Xu, J.; Chen, S.; Rawat, R. S.;Fan, H. J. Adv. Energy Mater. 2016, 6, 1600221.
[9]. Bharti, B.; Kumar, S.; Lee, H.; Kumar, R. Sci. Rep. 2016, 6, 32355.
[10]. Tripathi, N.; Islam, S. S. Appl. Nanosci. 2017, 7, 125-129.
[11]. Yang, J. W.; Esconjauregui, S.; Xie, R.; Sugime, H.; Makaryan,T.; D’Arsie, L.; Arellano, D. L. G.; Bhardwaj, S.; Cepek, C.; Robertson, J. J.Phys. Chem. C 2014, 118, 18683-18692.



